A palavra amonoácidopatia é a junção de “aminoácido”
(uma molécula existente nos alimentos) com “patia” (que significa doença). Dessa
maneira, as aminoacidopatias são doenças que dizem respeito aos aminoácidos.
Falar e entender sobre os aminoácidos não é uma
tarefa fácil porque exige um bom conhecimento de bioquímica. Mas, vou tentar facilitar as coisas para que todos possam
entender o assunto.
Nosso corpo é formado de células. E há bilhões delas. Mas, para sobreviverem, elas precisam de proteínas. Algumas células produzem suas próprias proteínas.
Porém, outras não as produzem. Neste caso, as proteínas precisam vir do exterior do corpo,
mais exatamente, dos alimentos que ingerimos. Ocorre que essas proteínas são grandes
demais para entrarem nas células. Por causa disso, precisam ser quebradas, liberando
substâncias contidas em seu interior. E, para isso, precisam dos aminoácidos.
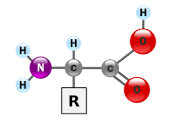
Os aminoácidos são moléculas formadas, basicamente, por quatro elementos-chaves: o carbono (C), o hidrogênio (H), o nitrogênio (N) e o oxigênio (O). Estes elementos se
juntam formando uma estrutura denominada “cadeia aminoácida”.

A esta cadeia podem-se juntar outras moléculas.
Frente a uma proteína, essa cadeia ataca e a quebra provocando, como reação, a liberação de substâncias importantes. As cadeias aminoácidas possuem estruturas variáveis, o que concorre para inúmeras possibilidades de combinações.


Mas, durante a divisão celular no início da formação do embrião, um erro pode
acontecer. Pode enfraquecer as proteínas fazendo com que sua quebra seja
facilitada quando não devia, ou torna-las tão fortes e resistentes, que fica impossível
de serem quebradas pelos aminoácidos. Quando há esse erro metabólico, os aminoácidos
produzem um efeito tóxico cuja conseqüência principal é um déficit funcional. isto significa que pode haver um acúmulo ou falta das proteínas necessárias.
O sistema nervoso é o primeiro a receber os
efeitos dessa toxidade e a sofrer com ela. Fato que pode provocar encefalopatias
leves, moderadas e graves. Outras vezes, essa toxidade é progressiva,
manifestando seus efeitos bem mais tarde, meses ou anos depois do nascimento.
Vamos conhecer alguns desses distúrbios:
.jpg)
A TIROSINEMIA é um erro genético do metabolismo que afeta a tirosina, responsável pela fabricação da uréia, uma substância tóxica para o organismo. A uréia é filtrada pelos rins, compõe a urina e é expelida por ela. Quando a tirosina não libera a uréia, ela fica concentrada, degenerando os rins e o fígado onde é produzida.
A tirosinemia tem três tipos:
A TIROSINEMIA tipo I
.jpg)
A tirosinemia é a mais comuns. Seu erro metabólico ocorre no cromossomo 15q23-25 e mais de 34 mutações diferentes
puderam ser detectadas e mapeadas. Sua incidência é de 22 casos a cada 1846
nascimentos e as famílias franco-canadenses
e seus descendentes são os mais atingidos.
Ela é causada por uma
mutação genética e hereditária que faz com que haja uma deficiência de
fumarilacetoacetato hidrolase (FAH) na
parte final da quebra da tirosina. A ausência dessa substãncia faz com que outra substância, a maleilacetoacetato, se acumule no organismo provocando
um grande efeito tóxico que atinge o fígado.
O diagnóstico pode ser
realizado ainda no período pré- natal, através do exame do líquido amniótico. Mas, quando esse diagnóstico
não se realiza nessa época, nos primeiros meses após o nascimento, o bebê
apresenta vômitos, icterícia, problemas no fígado, hipoglicemia,
ascite, sangramento, anorexia e irritabilidade. Quanto mais tarde o diagnóstico é realizado, mais
complicam-se os efeitos comprometendo o fígado de tal modo que há necessidade
de transplante. As alterações neurológicas são definitivas e observáveis.
TIROSINEMIA do tipo II
Ao contrário da tirosinemia
do tipo I, as funções do fígado, dos rins e as concentrações de outros aminoácidos
são normais. Porém, ao longo do crescimento vão aparecendo lesões nas córneas e
na pele. Muitas dessas lesões podem ser cancerígenas.
TIROSINEMIA do tipo III
Este tipo foi descoberto
recentemente e, por enquanto, poucas pessoas foram diagnosticadas com este
tipo. Relatos afirmam que os pacientes
não apresentaram sintomas. Fígado e rins apresentam funções normais. Porém, disfunções
neurológicas foram detectadas e relatados, tais como: atrasos do desenvolvimento, convulsões, ataxia
intermitente e comportamentos autodestrutivos.
Uma dieta alimentar pobre em tirosina, fenilalanina e metionina é
recomendada como tratamento em todos os tipos de tirosinemia. Esse tratamento e a dieta duram a vida toda. Embora não cure, amenizam os efeitos e o retardo
mental pode ser evitado.
fontes:
NORA JJ, Fraser FC. GENÉTICA MÉDICA. 3ª edição. Rio
de Janeiro: Guanabara Koogan, 1991.
NUSSBAUM RL, MCINNES RR, Willard HF. THOMPSOM e THOMPSOM: GENÉTICA MÉDICA. 6ª edição. Rio de Janeiro:
Guanabara Koogan, 2002.



{kind=link}
.jpg){kind=link}